DIVERSES APPLICATIONS DES PROGRAMMES DE GEOMETRIE
MOLECULAIRE DE GERARD LANGLET
Claude Chachaty
��� Parmi les contributions majeures� de G�rard Langlet � la physico-chimie figurent� ses logiciels pour la repr�sentation spatiale des mol�cules � partir de donn�es cristallographiques (1).� J'ai eu� la chance de pouvoir b�n�ficier de la rare comp�tence de G�rard dans ce domaine alors j'avais depuis peu la responsabilit� du laboratoire de R�sonance Magn�tique Nucl�aire� (RMN) et de R�sonance Paramagn�tique Electronique (RPE) du C.E.A Saclay. L'un des probl�mes majeurs �tait en effet de disposer rapidement de logiciels pour l'exploitation des� donn�es exp�rimentales. Pour faciliter l'utilisation de ses programmes et nous apprendre � en cr�er d'autres, G�rard a donn� en 1978 des cours d'initiation � APL .Ces cours ont �t� suivis avec un tel enthousiasme par les chercheurs du laboratoire, que nous avons adopt�� APL comme unique langage de programmation jusqu'au d�but de cette d�cennie en d�pit de quelques limitations techniques qui n'existent plus aujourd'hui.
��� Le but de ma collaboration avec G�rard �tait de cr�er des programmes APL pour d�terminer � partir de donn�es exp�rimentales de RMN, les vitesse de r�orientation des mol�cules en solution, leurs conformations pr�f�rentielles et les vitesses d'�change entre ces conformations. Pour mieux comprendre la nature de ces probl�mes, il me semble n�cessaire de mentionner au pr�alable quelques notions de base.
��� Les noyaux atomiques poss�dent pour la
plupart un moment magn�tique ![]() �associ� � leur spin et
sont donc assimilables � des aimants microscopiques qui interagissent � courte
distance� par l'interm�diaire des
liaisons chimiques (couplage scalaire J) ou � longue distance � travers
l'espace (couplage dipolaire D). La circulation des �lectrons dans le champ
magn�tique statique
�associ� � leur spin et
sont donc assimilables � des aimants microscopiques qui interagissent � courte
distance� par l'interm�diaire des
liaisons chimiques (couplage scalaire J) ou � longue distance � travers
l'espace (couplage dipolaire D). La circulation des �lectrons dans le champ
magn�tique statique ![]() �d'un spectrom�tre de
RMN g�n�re par ailleurs un champ de sens oppos� qui s'exerce sur les spins
nucl�aires et d�place les raies de r�sonance (d�placement diamagn�tique
s).
Le couplage dipolaire entre deux noyaux i et j est de la forme :
�d'un spectrom�tre de
RMN g�n�re par ailleurs un champ de sens oppos� qui s'exerce sur les spins
nucl�aires et d�place les raies de r�sonance (d�placement diamagn�tique
s).
Le couplage dipolaire entre deux noyaux i et j est de la forme :
�������������
�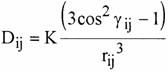 ����������� [1]
����������� [1]
o� ![]() �est l'angle entre le
vecteur
�est l'angle entre le
vecteur ![]() �joignant les deux noyaux
et le grand axe
�joignant les deux noyaux
et le grand axe ![]() de la mol�cule assimil�e � un ellipso�de et K, une constante
d�pendant de la nature des noyaux. Alors que les param�tres J et
s sont
directement mesurables sur le spectre de RMN, les couplages dipolaires qui contiennent
l'essentiel des informations sur la g�ometrie mol�culaire, ne peuvent �tre
obtenus dans les liquides� que par des
exp�riences de relaxation� nucl�aire.
L'exp�rience de relaxation la plus courante consiste � mesurer le temps de
relaxation longitudinale T1 qui est la constante de temps
d'alignement sur la direction de
de la mol�cule assimil�e � un ellipso�de et K, une constante
d�pendant de la nature des noyaux. Alors que les param�tres J et
s sont
directement mesurables sur le spectre de RMN, les couplages dipolaires qui contiennent
l'essentiel des informations sur la g�ometrie mol�culaire, ne peuvent �tre
obtenus dans les liquides� que par des
exp�riences de relaxation� nucl�aire.
L'exp�rience de relaxation la plus courante consiste � mesurer le temps de
relaxation longitudinale T1 qui est la constante de temps
d'alignement sur la direction de ![]() , de l'aimantation nucl�aire macroscopique
, de l'aimantation nucl�aire macroscopique ![]() �r�sultant d'un
ensemble de moments magn�tiques nucl�aires
�r�sultant d'un
ensemble de moments magn�tiques nucl�aires ![]() . Dans le cas le plus simple d'une mol�cule rigide
quasi-sph�rique en r�orientation rapide, la vitesse de relaxation d'un noyau i
en interaction dipolaire avec des noyaux j prend la simple forme :
. Dans le cas le plus simple d'une mol�cule rigide
quasi-sph�rique en r�orientation rapide, la vitesse de relaxation d'un noyau i
en interaction dipolaire avec des noyaux j prend la simple forme :
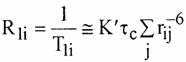 ����������������� [2]
����������������� [2]
o� ![]() �est le temps de
corr�lation qui correspond � peu pr�s ��
la dur�e moyenne de rotation� de
30� d'une mol�cule.
�est le temps de
corr�lation qui correspond � peu pr�s ��
la dur�e moyenne de rotation� de
30� d'une mol�cule.
��� En g�n�ral, les mol�cules organiques en solution ont plusieurs conformations, certaines pouvant poss�der un tr�s grand nombre d'isom�res rotationnels. On verra que les traitements informatiques des r�sultats experimentaux valables pour ces derni�res que nous qualifierons de flexibles, diff�rent notablement de ceux applicables aux �mol�cules semi-rigides o� les rotations internes ne s'effectuent qu'autour de deux ou trois liaisons.
��� Notre premi�re �tude commune concernait les conformations de� mol�cules semi-rigides comme les nucl�otides, en particulier l'ad�nosine 5' monophosphate (AMP). Le probl�me nous avait �t� pos� par une �quipe de l'Institut Pasteur de d�terminer le changement de conformation de ce nucl�otide lorsqu'il se fixe sur le site r�cepteur d'une enzyme, la phosphorylase b, dont il est activateur (2). L'AMP comporte un cycle ribose li� � la base ad�nine compos�e d'un cycle pentagonal et d'un cycle hexagonal et � un groupe phosphate (figure 1). Les probabilit�s des deux conform�res N et S du cycle ribose et des trois rotam�res du groupe phosphate se d�duisent simplement des couplages J entre protons vicinaux. Il n'existe par contre� aucun couplage J entre les protons du cycle ad�nine et ceux du cycle ribose de sorte que les orientations relatives des deux cycles ne peuvent �tre d�duites que� des vitesses de relaxation de ces protons qui d�pendent de� leurs distances mutuelles (equ. 2).
��� Ce probl�me a �t� r�solu gr�ce au syst�me GEOMOL de G�rard, un groupe de programmes qui permet de calculer ab initio ou � partir de donn�es cristallographiques, les coordonn�es atomiques pour n'importe quelle conformation en choisissant les angles de torsion entre diff�rentes liaisons. Le programme FIG de ce groupe dessine les formes mol�culaires produites, avec �ventuellement des effets de perspective comme sur la figure 1 ou la repr�sentation des atomes par des sph�res dont le rayon correspond � leur distance minimale d'approche (rayon de Van der Waals).
��� Pour d�terminer les conformations les plus probables de l'AMP on calcule tout d'abord les distances entre un proton isol� de la base et les trois protons les plus proches du cycle ribose en fonction de l'angle de rotation Y autour de leur liaison commune.
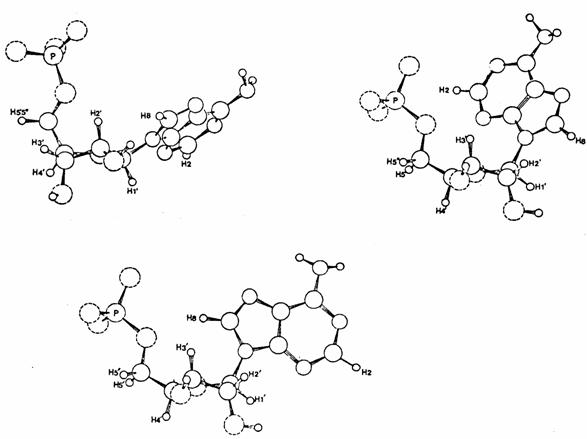
Figure 1. Conform�res pr�ferentiels de l'ad�nosine 5' monophosphate repr�sent�s comme pour les figures suivantes, � l'aide du programme FIG de G�rard Langlet.
���� Les deux valeurs les plus probables ![]() �et
�et ![]() �de
Y
associ�es aux formes N et S du cycle ribose correspondent � l'�cart-type
minimum entre les vitesses de relaxation exp�rimentales et celles calcul�es par
un programme annexe utilisant la relation [2] pour les quatre protons en
question. Cette m�thode est d�crite� en
d�tail dans les r�f�rences 3 et 4.
�de
Y
associ�es aux formes N et S du cycle ribose correspondent � l'�cart-type
minimum entre les vitesses de relaxation exp�rimentales et celles calcul�es par
un programme annexe utilisant la relation [2] pour les quatre protons en
question. Cette m�thode est d�crite� en
d�tail dans les r�f�rences 3 et 4.
��� Au moment o� ce travail a �t� effectu�, nous ne disposions que de zones de 80 Ko. Le tour de force de G�rard Langlet a �t� d'�crire des programmes tr�s performants et d'utilisation assez simple, dans un espace aussi r�duit.
��� A partir de 1980, notre collaboration a port� plus particuli�rement sur l'�tude de mol�cules flexibles comportant g�n�ralement une cha�ne hydrocarbon�e. Il s'agissait de mol�cules utilisables pour l'extraction d'ions m�talliques et dont certaines sont des agents tensioactifs formant des micelles et des cristaux liquides.
��� Selon le mod�le d'isom�rie rotationnelle de Flory (5), chaque groupe de trois liaisons successives d'une telle mol�cule, poss�de trois rotam�res :� trans (T), gauche+ (G) et gauche- (g) repr�sent�s sur la figure 2.
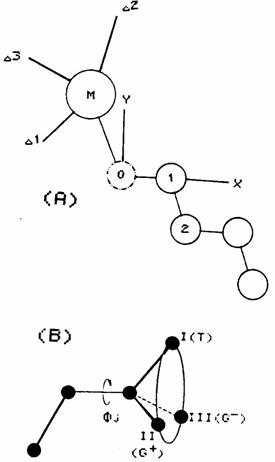
Figure 2. (A)
Syst�me de r�f�rence mol�culaire. Dans ce r�f�rentiel, seuls les atomes 0 et 1
de la cha�ne sont fixes, M est un ion m�tallique dans un complexe de
coordination ou l'atome central de la t�te polaire d'une mol�cule tensioactive.
Les axes ![]() �forment un r�f�rentiel
secondaire choisi en fonction syst�me �tudi�. (B) D�finition des rotam�res
autour d'une liaison de la cha�ne.
�forment un r�f�rentiel
secondaire choisi en fonction syst�me �tudi�. (B) D�finition des rotam�res
autour d'une liaison de la cha�ne.
��� Pour une cha�ne � N atomes, le plus souvent
des carbones, il existe donc potentiellement ![]() ��ou
��ou ![]() �conform�res selon que
l'on prend ou non en compte l'atome M auquel est li� la cha�ne, dont certains sont
interdits st�riquement. Comme dans le cas pr�c�dent, le calcul des vitesses de
relaxation ou des d�placement des raies de r�sonance nucl�aires n�cessite au
pr�alable celui des coordonn�es des atomes dans un syst�me de r�f�rence
mol�culaire pour toutes les conformations permises. Dans ce but G�rard Langlet
a �crit le programme CONSTRUC,
remarquablement facile � utiliser. Chaque atome d'une cha�ne �tant d�sign�
selon sa nature par une lettre, on d�finit tout d'abord les longueurs des
liaisons entre les atomes et les angles entre deux liaisons successives. Pour
g�n�rer les coordonn�es des atomes, il suffit d'entrer ensuite leur s�quence
dans la cha�ne comme le montre l'exemple le plus simple d'une cha�ne � quatre
atomes, celle de la propylamine :
�conform�res selon que
l'on prend ou non en compte l'atome M auquel est li� la cha�ne, dont certains sont
interdits st�riquement. Comme dans le cas pr�c�dent, le calcul des vitesses de
relaxation ou des d�placement des raies de r�sonance nucl�aires n�cessite au
pr�alable celui des coordonn�es des atomes dans un syst�me de r�f�rence
mol�culaire pour toutes les conformations permises. Dans ce but G�rard Langlet
a �crit le programme CONSTRUC,
remarquablement facile � utiliser. Chaque atome d'une cha�ne �tant d�sign�
selon sa nature par une lettre, on d�finit tout d'abord les longueurs des
liaisons entre les atomes et les angles entre deux liaisons successives. Pour
g�n�rer les coordonn�es des atomes, il suffit d'entrer ensuite leur s�quence
dans la cha�ne comme le montre l'exemple le plus simple d'une cha�ne � quatre
atomes, celle de la propylamine :
ATOMES� :� N������
C������ C������ C
ANGLES� :������� 120.00 �110.00
CHI (c) est
l'angle entre les plans� N-3, N-2, N-1 et
N-2, N-1, N form�s par les atomes. Les r�sultats de CONSTRUC sont regroup�s
dans le tableau :
�������������
Tableau 1
��������������
Atomes� Conformations�����������������
Coordonn�es�������������������
��� Le programme CONSTRUC ne donne que les coordonn�es des atomes constituant la cha�ne. Celles des atomes� "p�riph�riques" tels que les hydrog�nes d'une cha�ne hydrocarbon�e, s'en d�duisent par des op�rations simples.
�� Dans le r�f�rentiel x,y,z de la figure 2,
les trois positions possibles pour l'atome 2 correspondent aux rotam�res T, G
et g de la s�quence des atomes M, 0, 1, 2;��������������������������������
le nombre de positions que peut occuper le dernier atome de la cha�ne est �gal au nombre de conformations de celle-ci, soit 9 pour l'exemple du tableau 1. Dans ce tableau, les coordonn�es ont �t� calcul�es pour l'option 1 du programme, valable pour une g�om�trie standard avec c = 0, 120�, -120� autour de chaque liaison. L'option �1 permet de choisir pour n'importe quelle liaison c = a, a+b, a-b, a etb �tant des angles quelconques. Avec cette seconde option, il devient� possible de construire pratiquement toute mol�cule lin�aire avec comme seule limite un nombre de conformations compatible avec la taille de la zone de travail. Pour une zone de 15 Mo, cette limite se situe� entre 300 et 500 selon le probl�me �tudi�.
��� Toutes
les observables RMN que nous nous proposons de calculer (d�placement
diamagn�tique ou paramagn�tique, vitesses de relaxation) sont du type dipolaire
et d�pendent lin�airement de termes en ![]() , o� D est de la� forme
de la relation [1]. Ce sont des moyennes pond�r�es par les probabilit�s des
conformations que l'on calcule � partir du vecteur PT des probabilit�s� du rotam�re trans autour des liaisons 0_1, 1_2,...,N-2_N-1. En supposant pour
simplifier que les rotam�res G et g sont �quiprobables, on obtient les vecteurs
de probabilit�s PG�Pg
�0.5�1-PT.
Partant du tableau CONF des
conformations mol�culaires, on calcule le vecteur de probabilit�s PROB de
toutes les conformations, par les op�rations :
, o� D est de la� forme
de la relation [1]. Ce sont des moyennes pond�r�es par les probabilit�s des
conformations que l'on calcule � partir du vecteur PT des probabilit�s� du rotam�re trans autour des liaisons 0_1, 1_2,...,N-2_N-1. En supposant pour
simplifier que les rotam�res G et g sont �quiprobables, on obtient les vecteurs
de probabilit�s PG�Pg
�0.5�1-PT.
Partant du tableau CONF des
conformations mol�culaires, on calcule le vecteur de probabilit�s PROB de
toutes les conformations, par les op�rations :
dim�(3*N-2),N-2
Ptrans�(CONF='T')�dimrPT
Pgauche
�0.5�((CONF='G')+CONF='g')�dimr1-PT
PROB��/Ptrans+Pgauche
�� En reprenant l'exemple du tableau 1 avec PT�0.7 0.5 on obtient ainsi :
Tableau 2
��� CONF��������������� Ptrans�������������������������� Pgauche� ������������������������PROB
TT������� 0.7 0.5�������� 0.00 0.00�������� 0.3500
gg�������
0.0 0.0�������� 0.15 0.25�������� 0.0375
��� Les contraintes st�riques sont prises en compte en �liminant les conformations o� les distances entre atomes non li�s sont inf�rieures � la somme de leur rayon de Van der Waals, ce qui a pour effet de r�duire consid�rablement leur nombre. Ces� m�thodes� mises en oeuvre dans une s�rie de programmes dont CONSTRUC est le noyau commun, sont expos�es en d�tail dans la r�f. (6). Voici quelques exemples de leurs applications :
Complexes
de coordination paramagn�tiques.
��� Les mol�cules poss�dant des groupes
polaires entre autre les amines, les alcools ou les phosphates ont la propri�t�
de fixer des ions m�talliques dont certains sont paramagn�tiques car ils
poss�dent au moins un spin �lectronique non appari�. Le moment magn�tique
correspondant est de trois ordres de grandeur sup�rieur � ceux des moments nucl�aires.
Il exerce un champ intense sur les noyaux voisins dont il augmente les vitesses
de relaxation et d�place les raies de r�sonance. Ces propri�t�s sont� mises � profit pour des applications de la
RMN au conformations des mol�cules en solution (7). La
figure 3 repr�sente le complexe de coordination qui� a servi de mod�le pour la mise au point de
nos programmes.
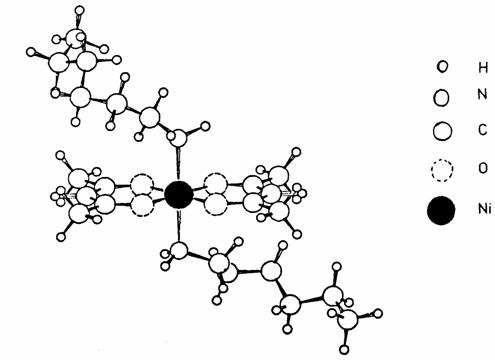
Figure 3. Complexe de l'hexylamine avec l'ac�tylac�tonate de Nickel.
Carot�noporphyrines
��� Le champ magn�tique induit par la circulation des �lectrons dans un cycle aromatique peut �tre assimil� � celui provenant d'un dip�le magn�tique en son centre et d�place les r�sonances des noyaux voisins. Cet effet a �t� utilis� pour d�terminer les conformations des carot�noporphyrines poss�dant une cha�ne caroteno�de li�e � un macrocycle porphyrine qui comporte 12 dip�les de ce type. Les carot�noporphyrines servent � �tudier les transfert d'�nergie entre la chlorophylle et le carot�ne qui interviennent en photosynth�se.
|
|
|
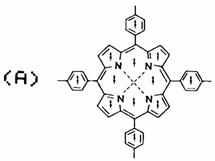
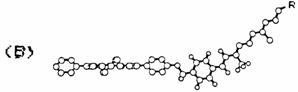
Figure 4. (A) Dip�les magn�tiques cr��s par les courants de cycle de la t�traph�nyl-porphyrine. (B) Conformation d'une carot�noporphyrine� d�termin�e par RMN du proton (8).
Micelles
et cristaux liquides
��� Au del� d'une concentration critique, les mol�cules tensioactives� forment au contact de l'eau des agr�gats sph�riques, les micelles (figure 5). En solution concentr�e, les micelles peuvent fusionner pour donner des phases cristallines liquides. Ces diff�rents stades impliquent des changements dans les� conformation et la� dynamique des mol�cules tensioactives qui tendent � s'orienter perpendiculairement � l'interface de ces agr�gats avec l'eau en occupant un volume minimum. La relaxation nucl�aire est la m�thode la plus sp�cifique pour� �tudier ces ph�nom�nes. Nous avons utilis� la relaxation� du carbone 13 induite soit par un ion paramagn�tique � l'interface eau/agr�gat, soit par les protons de la cha�ne hydrocarbon�e (9).
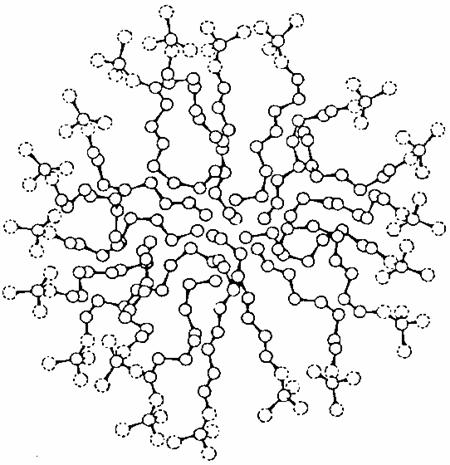
Figure 5. Micelle d'octylphosphate de sodium.
��� Le programme CONSTRUC a �t� �galement utilis� dans une �tude� r�cente sur l'int�raction d'�change entre deux spins �lectroniques localis�s � chaque extr�mit� de la cha�ne flexible d'un biradical nitroxyde (10). Le param�tre d'�change moyenn� sur toutes les conformations est ajust� de fa�on � reproduire au mieux le spectre de RPE du biradical.
���� Ces quelques exemples montrent la diversit� des applications de programmes� cr��s au cours d'une dizaine d'ann�e de collaboration avec G�rard Langlet dont certains sont encore utilis�s. Je crains que G�rard n�ait pas sauvegard� le syst�me GEOMOL et les programmes qu�il contient pour dessiner les mol�cules. J�ai par contre conserv� le programme CONSTRUC avec ses sous-fonctions, qui sont list�s en annexe.
��� Ma� collaboration avec G�rard s'effectuait sous forme de discussions informelles, g�n�ralement assez� br�ves. En plus de son talent d'informaticien, il avait en effet une connaissance approfondie de la physico-chimie qui lui permettait de saisir rapidement� tous les aspects d'un probl�me que je lui exposais. Il m'a par ailleurs aid� � utiliser efficacement� APL en� me donnant des conseils pour r�duire le temps et l'espace n�cessaires � l'ex�cution des programmes. De tous les chercheurs que j'ai rencontr�, c'est incontestablement G�rard qui a eu l'influence la plus positive sur mon travail et je lui en suis tr�s reconnaissant.
R�f�rences
1� G. Langlet
Journal of Applied Crystallography, 5, 66 (1977).
2� M. Morange, A. Kolb, H. Buc, C. Chachaty et G. Langlet
�European Journal of� Biochemistry, 74, 99 (1977).
3� C. Chachaty et G. Langlet
�FEBS Letters, 68, 181 (1976).
4� C. Chachaty, B. Perly, A. Forchioni et G. Langlet
Biopolymers, 19, 1211 (1980).
5�
P.J. Flory,
Statistical Mechanics of Chain Molecules, Interscience, New-York 1969
6� C. Chachaty et G. Langlet
Journal de Chimie Physique, 82, 613 (1985).
7� C. Chachaty, B. Perly et G. Langlet
�Journal of� Magnetic Resonance, 50, 125 (1982).
8�
C. Chachaty, D. Gust, T.A. Moore,
G.A. Nemeth, P.A. Liddel et A.L. Moore
�Organic Magnetic Resonance, 22,
39 (1984).
9� C. Chachaty et Th. Bredel
��Journal of the Chemical
Society, Faraday Transactions, 88, 1893 (1992).
10 C. Chachaty, S. Gambarelli et A. Rassat
�Magnetic Resonance in Chemistry, �33, S174 (1995).
Programmes de G�rard Langlet pour la g�om�trie
des mol�cules � cha�ne flexible.
��� Le programme CONSTRUC calcule les coordonn�es des atomes pour la forme initiale de la cha�ne contenue dans le plan XY de la figure 2 puis, � l�aide de la fonction ROTTI, effectue ce m�me calcul pour tous les isom�res possibles de cette cha�ne en effectuant des rotations autour des liaisons successives.
CONSTRUC;S;A;B;C;L;Q;VL;VA;n;ŒIO;chi;D;K
ŒIO„0 � A„'HCNOSPFc',0�B„'-�=�',0�b„'1A23',0�n„'non
defini:'
E:…(3>�S„S‡�,0�S„��„'CHAINE?')/E
–(0�L„S�C„A,B)/'…E,�Œ„''INTERDIT
: '',(~L)/S'
S„(S�' ')/S„,S,[0.5]'
-'[(�1‡L^1�L„S�A),0]
Z[L/��S]„b[B�(L„S�B)/Z„S]
Z„((�Z)� 1 0)š� 0 1 2 3
4 �(5,�Z)�Z
–(1�N„2�ŒNC Q„
�2 0 ‡Z)/'…0,�Œ„n,,'' '',NšQ'
'ATOMES : ',6‡,(((�AT),7)�'
'),AT„(S�A)/S
'NUMEROS : ',6‡ 8 0 •��AT
'ANGLES �: �����', 8 2 •-VA„((�VA)� �1 1)�VA„–,',',Q
–(1�N„2�ŒNC Q„ �1 �2 ‡Z)/'„0,�Œ„n,,''
'',N�Q'
'LIAISONS : ', 8 2 •VL„–,',',Q
Z„0,[0]+™(K,[0.5]-K„VL�(�VL)� �1 1)׳ 2 1
�.��-\1,|VA�180
chi„((D„�2+1†�Z)�0)�.+ 0 120 �120
'Taper 1 pour CHI standard (0 120 �120)' � …('1'=1†,�)/EC
ER:'ENTRER ',(•3�D),' VALEURS'
–((3�D)��/�chi„Œ)/'…ER,�Œ„''Nb. de
valeurs incorrect'''
chi„�(D,3)�chi
EC:Z„(CHI„chi)ROTTI Z,0
'Coordonn�es : r�sultat dans Z. Dimensions : ',�Z
b„�Z � QFR11„˜0.5+10000�Z
R„CHI ROTTI
T;G;H;K;U;D;M;N;Q;CH;L;C;W;ŒIO
ŒIO„0
�ROTATION CHI SUR TABLEAU T (ATTENTION : SENS INVERSE)
–(2���CH„-�CHI�180)/'�R„0�Œ„''CHI : ERREUR'''
–((3>N„1†�T)Ÿ 3 2 Ÿ.�(�1†�T),��T)/'…�R„0�Œ„''T
. ERREUR'''
�ON FAIT TOURNER DE CHI (CHI DOIT ETRE UN TABLEAU EN �
�LE DERNIER POINT (N) AUTOUR DE (N-2),(N-1)
�ETC... JUSQU'AU POINT 2 QUI TOURNE AUTOUR DE (0,1)
–((�2+N)�1†�CHI)/'…R„0�Œ„''CHI doit
avoir 2 lignes de moins que T'
G„1†�L„�L�((+/L�L„(1
0 ‡ �1 0 ‡T)- �2 0 ‡T)*0.5)�.+3�0
M„((2 0 2 1 �L�.�L)�.+K[1]�0)�(3
3 ,K„�C)�1-C„2�CH
Q„C,[0] 1 0 1 2 �(L,-L)�.�S„1�CH
M„M+(3 3 ,�C)�Q[0 3 5 6 0 1 2 4
0 ;;] � H„2†N-1 � W„�T
E:R„ŒEX 'R'
T„T-W�T[H[0]-2;] � R„(H‡T)+.�M[;;0;]
� M„ 0 0 1 0 ‡M
D„�R„ 0 2 1 �R � R„((�/2†D),2‡D)�R
� W„�T„((0 3 +H)†T),[0]R
…E—�2ˆ1†H„H- 1 0 � R„T
��� La fonction FTGg �construit le tableau de
caract�res qui donne les rotam�res T,
G et g �autour des liaisons 0_1,
1_2,...,N-2_N-1 o� N est le nombre des atomes de la
cha�ne, correspondant au tableau des coordonn�es (voir tableau 1 et figure 2).
FTGg N;ŒIO;K;j;B;D
–(N^.� 3 4 5 6 7 8 9)/'…0,�Œ„''NON! SCAL. DE 3 A
9'''
D„1‡�TCH„ 0 1 �' '
E:TCH„(D† 1 0 •N),[ŒIO„0]TCH � K„�B„,�(3,�B)�B„TCH[;0]
B„(K�B),[0.5]K�'TGg' � j„�TCH„ 0 1 ‡TCH
D„�TCH„(K,1‡j)� 1 0 2 �(3,j)�TCH
D„1‡�TCH„(0 �2 -D)†TCH � TCH[; 0 1]„B � B„ŒEX
'B'
…(1<N„N-1)/E � TCH„((2,D)†
2 1 �'01'),[0]TCH
�����
��� La fonction Gg indique les occurences des formes locales Gg ou gG pour �liminer
�ventuellement les conformations�
st�riquement d�favorables qui en contiennent (cf. r�f. 5). Cette fonction
ne s�applique pas � la liaison 0_1, non plus qu�� la liaison N-2_N-1 si la
cha�ne se termine par un groupe m�thyle (option METH=1).
Gg;B;j;ŒIO;A
�FABRIQUE LE TABLEAU BINAIRE Ng (G OU g) ET LE VECTEUR NGg
�(NOMBRE DE Gg OU DE gG)
–(2�ŒNC 'TCH')/'…0,�Œ''FAIRE EN PREMIER . FTGg'''
TCH„ 0 1 ‡TCH � A„'G'=
0 �1 ‡TCH � j„'g'= 0 1 ‡TCH � A„A^j �j„ŒEX 'j'
B„'g'= 0 �1 ‡TCH � j„'G'=
0 1 ‡TCH � B„B^j � j„ŒEX 'j'
A„AŸB
�MODIF DU 2/12/86 POUR EVITER PRISE EN COMPTE DE LIAISON 0-1
A„ 0 1 ‡AŸB � B„ŒEX
'B' � –(METH�1)/'NGg„+/A'
�POUR EVITER ELIMINATION DE Gg DUE AU METHYLE TERMINAL
–(METH=1)/'NGg„+/(0 �1‡A)'
� A„ŒEX 'A' � Ng„TCH�'Gg'
��� Les fonctions FTGg et Gg �sont utilis�es pour d�terminer le vecteur de probabilit�s des conform�res comme expliqu� dans le texte (voir �galement le tableau 2).